- Article
- Source: Campus Sanofi
- 23 Oct 2023
ODYSSEY OUTCOMES Safety Profile Praluent® (alirocumab)

Odyssey outcomes safety profile consistent with that of Praluent® (alirocumab) phase 3 placebo-controlled trials1
Praluent® safety and tolerability profile
Patients treated with Praluent® experienced a similar frequency of adverse events to those on placebo, but with more local injection site reactions* (p<0.001) — although these were usually mild and self-limited.2
No difference in safety profile was observed between the two doses of Praluent® (75 mg and 150 mg) used in the phase 3 program.1†
Adverse events2
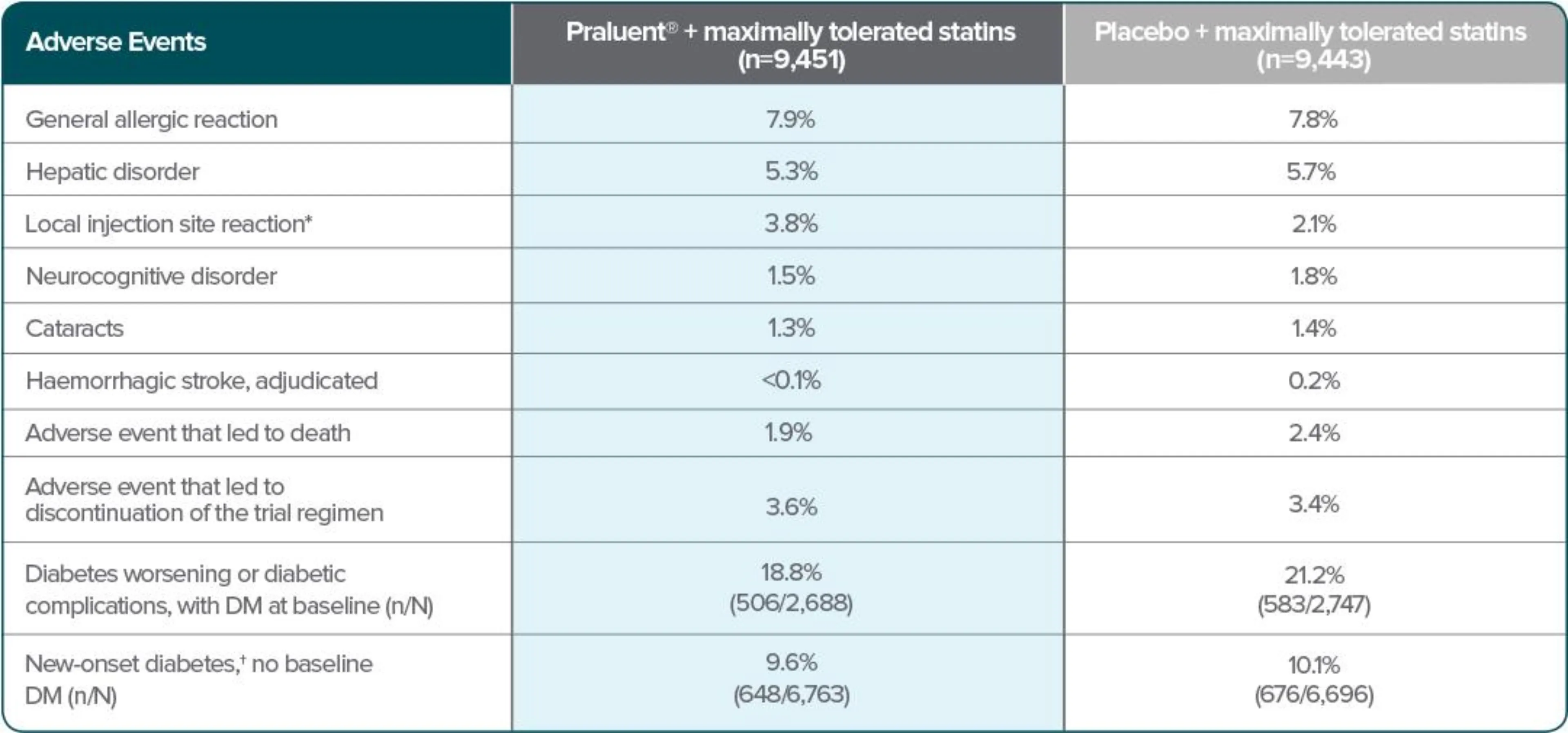
Adapted from Schwartz et al. 2018.
*p<0.001
†New-onset diabetes was defined according to the presence of one or more of the following, with confirmation of the diagnosis by blinded external review by experts in the field of diabetes: an adverse event report, a new prescription for diabetes medication, a glycated hemoglobin level of at least 6.5% on two occasions (and a baseline level of <6.5%), or a fasting serum glucose level of at least 126 mg/dL on two occasions (and a baseline level of <126 mg/dL).2
DM=diabetes mellitus
Additional safety information
Contraindications1
Hypersensitivity to the active substance or to any of the following excipients: Histidine, Sucrose, Polysorbate 20, water for injection.
Summary of safety profile1
In ten phase 3 controlled trials, involving patients with primary hypercholesterolemia and mixed dyslipidaemia, the most common adverse reactions were local injection site reactions, upper respiratory tract signs and symptoms, and pruritus. Most common adverse reactions leading to treatment discontinuation in patients treated with Praluent® were local injection site reactions.
Immunogenicity/anti-drug-antibodies (ADA)1
Anti-drug antibody responses, including NAb, were low titre and did not appear to have a clinically meaningful impact on the efficacy or safety of Praluent®, except for a higher rate of injection site reactions in patients with treatment-emergent ADA compared to patients who were ADA negative (7.5% vs 3.6%). The long-term consequences of continuing Praluent® treatment in the presence of ADA are unknown.
In a pool of 10 placebo-controlled and active-controlled trials of patients treated with Praluent® 75 mg and/or 150 mg Q2W as well as in a separate clinical study of patients treated with Praluent® 75 mg Q2W or 300 mg Q4W (including some patients with dose adjustment to 150 mg Q2W), the incidence of detecting ADA and NAb was similar to the results from the ODYSSEY OUTCOMES trial.
LDL-C values <25 mg/dL (<0.65 mmol/L)
Although adverse consequences of very low LDL-C were not identified in alirocumab trials, the long-term effects of very low levels of LDL-C are unknown.
*Injection site reactions included itching, redness or swelling.2
†In ten phase 3 controlled trials, involving patients with primary hypercholesterolemia and mixed dyslipidaemia, the most common adverse reactions were local injection site reactions, upper respiratory tract signs and symptoms, and pruritus. Most common adverse reactions leading to treatment discontinuation in patients treated with Praluent® were local injection site reactions.1
Praluent®
Find more information on Indication, Administration and Mechanism of Action and watch videos about Praluent®.
.png)
References
ADA=anti-drug antibodies; LDL-C=low-density lipoprotein cholesterol; NAb=neutralizing antibodies; Q2W=once every two weeks; Q4W=once every four weeks.
1. Praluent Summary of Product Characteristics. Available at https://www.medicines.org.uk/emc/product/8093/smpc. Accessed October 2023.
2. Schwartz G, et al. N Engl J Med 2018;379:2097–2107.
MAT-XU-2204592 (v3.0) Date of Preparation: October 2023